
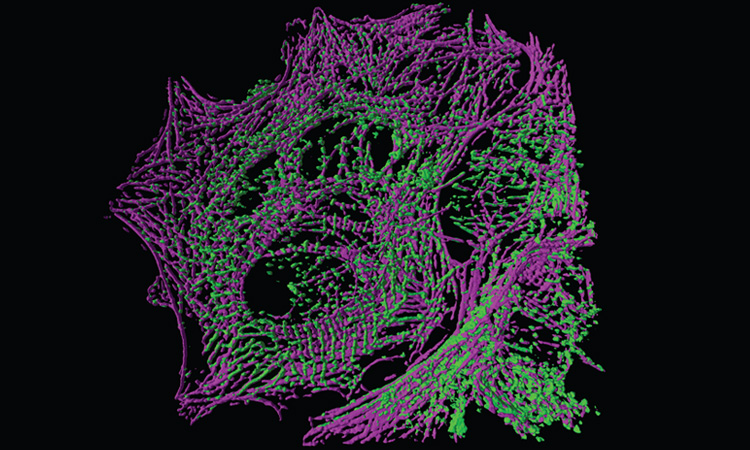
 Rendering of a cardiomyocyte, with actin shown in magenta and alpha-actinin in green, as imaged by oblique-plane structured-illumination microscopy (OPSIM). [J.B. Hayes, Vanderbilt Univ. / R. Fiolka, F. Zhuo, UT Southwestern Med. Ctr.]
Rendering of a cardiomyocyte, with actin shown in magenta and alpha-actinin in green, as imaged by oblique-plane structured-illumination microscopy (OPSIM). [J.B. Hayes, Vanderbilt Univ. / R. Fiolka, F. Zhuo, UT Southwestern Med. Ctr.]
In the late 1850s, German entrepreneur Carl Zeiss faced a dilemma. His Jena workshop had started building simple, low-power microscopes only a few years earlier, moving away from other types of scientific tools and instruments. But now, scientists and medical professionals were demanding higher-magnification, compound microscopes—the design of which, at the time, was based on laborious trial and error, with no mathematical foundation.
To get to a more efficient, scientific process, Zeiss recruited a young physicist, Ernst Abbe, to work on the problem. And Abbe had a groundbreaking realization: Despite the workshop’s best efforts, a microscope’s resolution would always hit a wall due to the diffraction of light. He published in 1873 what is today known as Abbe’s diffraction limit, which states that two illuminated objects can be resolved only if they are separated by a distance greater than about half the wavelength of light.
For more than 100 years, Abbe’s diffraction limit stood the test of time, despite attempts to overcome it with techniques like confocal and multi-photon microscopy. Finally, in 2000, the Romanian-German physicist Stefan Hell ingeniously got around this barrier with his first experimental demonstration of stimulated emission depletion (STED) microscopy. Hell had conceptualized STED as a postdoc years earlier, stirring up much controversy—but STED images of baker’s yeast and the bacterium E. coli revealed that the idea actually worked. “The diffraction barrier responsible for a finite focal spot size and limited resolution in far-field fluorescence microscopy has been fundamentally broken,” the paper boldly announced.
The super-resolution revolution has given biologists an unprecedented look inside cells with light—and researchers continue to push the technique’s boundaries.
Later, Eric Betzig and William Moerner, working separately, laid the foundation for another approach to imaging beyond Abbe’s limit. In 2006, Betzig reported the use of single-molecule localization microscopy (SMLM)—enabled by Moerner’s discovery that the fluorescence from individual molecules can be controlled by light—to image lysosome membranes at a resolution far better than the diffraction limit. Eight years later, Hell, Betzig and Moerner shared the 2014 Nobel Prize in Chemistry for their development of an entirely new optical-imaging category: super-resolution microscopy.
Since then, the super-resolution revolution has given biologists an unprecedented look inside cells with light, at a level of detail fine enough to see subcellular and suborganelle phenomena—previously only achievable with electron microscopy. And researchers continue to push the boundaries of the technique, inventing new imaging mechanisms and further improving spatial and temporal resolution.
“In the next 5 to 10 years, super-resolution microscopy techniques will head for application, and they are expected to be used in more research fields,” says Jia Li of the College of Physics and Optoelectronic Engineering at Shenzhen University in China. The technique, Li adds, should enable exploration of subcellular structures and their relationships, and may even provide new insights on molecular targets for treating autoimmune and neurodegenerative diseases.
End runs around Abbe’s diffraction limit
At the Friedrich Schiller University in Jena, a large stone sphere on the campus is etched with Abbe’s famous expression for the minimum distance, d, that can be resolved by an optical microscope: d = λ /(2n sin α), where λ is the wavelength of the observed light, n is the refractive index of the immersion medium and α is the angle of the cone over which the objective can capture light from the sample.
In practical terms, d typically ranges from 200 nm for far-blue light to 350 nm for far-red light. This meant scientists could observe the contours of some cellular organelles, like mitochondria. But anything smaller—viruses; the exact distribution of proteins and small molecules; fine-scale cell structures such as neural synapses—could not be resolved with light.
The first technique to go meaningfully beyond Abbe’s diffraction limit was STED, proposed by Hell in 1994. STED uses two lasers, one for exciting fluorescence in the sample and an additional laser with an annular (donut-shaped) beam. The annular laser is used to quench all fluorescence except that of a nanometer-sized volume in the middle—the “donut hole.” The beams sweep across the sample, creating a comprehensive image out of the tiny, nanometer-sized donut holes. The sub-diffraction resolution hinges on the efficiency of the stimulated emission process, which depends strongly on the depletion-beam intensity.
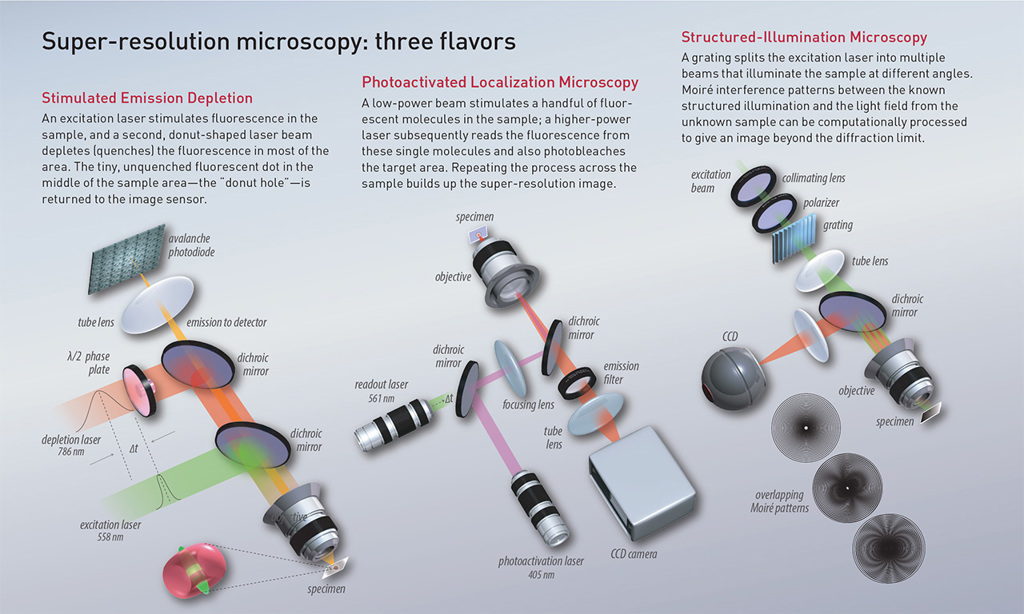 [Illustration by Phil Saunders] [Enlarge graphic]
[Illustration by Phil Saunders] [Enlarge graphic]
SMLM—which actually refers to a family of super-resolution techniques—uses localizations of individual molecules to build up an overall structure. Moerner’s revolutionary work on measuring single fluorophores effectively showed that, if a single molecule could be localized with high precision, so could two molecules very close to one another—as long as they could be made to fluoresce at different times.
Betzig leveraged the idea in photoactivated localization microscopy (PALM), in which a weak light pulse activates only a small fraction of fluorescent proteins in a sample. After bleaching or switching them off, a new subset can be activated. This process is repeated many times, and the resulting images are processed and superimposed to generate a single super-resolution image. Another SMLM approach, stochastic optical reconstruction microscopy (STORM), relies on the imaging of inherently or induced blinking organic dyes rather than fluorescent proteins.
Both STED and SMLM have achieved impressive results, with a 20-to-40-nm lateral and 70-nm axial resolution for STED and a 10-to-15-nm lateral and 50-nm axial resolution for SMLM. But both also have their drawbacks. STED’s high-intensity illumination can contribute to photobleaching and possible phototoxicity for living cells. And the temporal resolution of SMLM suffers from the technique’s need to acquire thousands of images for a single reconstruction—which makes it tough to capture the dynamics of living cells. But researchers continue to seek novel ways to move beyond those limitations and take super-resolution microscopy even further.
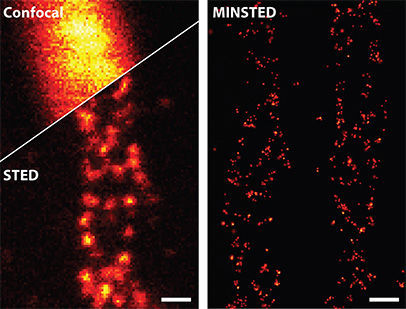 Fluorescently labeled proteins (Mic60) in a cell’s mitochondria, imaged with conventional confocal microscopy (top left), STED nanoscopy (bottom left) and MINSTED (right). [© MPI for Multidisciplinary Sciences / M. Weber; data: M. Weber et al., Nat. Photon. 15, 361 (2021)]
Fluorescently labeled proteins (Mic60) in a cell’s mitochondria, imaged with conventional confocal microscopy (top left), STED nanoscopy (bottom left) and MINSTED (right). [© MPI for Multidisciplinary Sciences / M. Weber; data: M. Weber et al., Nat. Photon. 15, 361 (2021)]
A different flavor of donut
Arguably, the biggest recent breakthrough in super-resolution microscopy once again originates from Hell’s laboratory, in a pair of novel techniques called MINFLUX (MINimal emission FLUXes) and MINSTED (MINimal STimulated Emission Depletion). By combining the strengths of STED and SMLM, MINFLUX and MINSTED provide localization precision at the scale of the fluorophore itself (1 to 3 nm), offering resolutions below 5 nm.
Initially reported in 2016, MINFLUX switches individual fluorophores on and off randomly, as in SMLM, and then determines their exact positions using a donut-shaped laser beam, similar to STED. For MINFLUX, however, the donut-shaped light is used for excitation rather than quenching. Fluorophores sitting on the donut portion will be excited, while those in the donut hole will not. A computer algorithm then lets MINFLUX pinpoint the coordinates of each fluorophore with molecule-size resolution.
“From a physics standpoint, achieving down to one nanometer localization precision of single fluorophores with MINFLUX has been an impressive feat,” says Lothar Schermelleh, with the University of Oxford biochemistry department. “While the meaningful biological applicability of MINFLUX is still rather limited, mainly to single-molecule tracking, within the next few years doing structural biology inside cells should become possible.”
The technique has other advantages over SMLM, such as a reduction in the number of photons needed to localize an emitter and an enhancement in temporal resolution, up to 100-fold. In 2022, Hell’s laboratory used a MINFLUX microscope to record—at 1.7-nm precision, and within less than a millisecond—the stepping motion of kinesin-1, a motor protein that transports cellular cargo along microtubules. The work demonstrated the potential for following single-molecule dynamics and making inferences on structural changes from datasets obtained with MINFLUX.
“MINFLUX enables substantially sub-millisecond time resolution in molecular tracking, thus providing statistically robust tracking data, with large numbers of samplings of position in time,” says Steffen J. Sahl, a longstanding collaborator of Hell’s and research scientist at the Max Planck Institute for Multidisciplinary Sciences, Göttingen. “The study of dynamical movements and even structural rearrangements measured with single dyes as reporters is being revolutionized.”
From nanometers to angstroms
The general principle behind MINFLUX has been extended to several other super-resolution microscopy methods. Repetitive optical selective exposure (ROSE), reported in 2019, localizes emitters by estimating their relative positions with respect to shifting interference fringes. A year later, researchers implemented the MINFLUX concept with sinusoidal illumination patterns to create SIMFLUX. Both techniques are readily compatible with imaging a large field of view and provide around a twofold improvement in the localization precision over conventional camera-based localization at the same photon budget. Modulation localization (ModLoc) microscopy, reported in 2019, employs an illumination modulation approach with shifting tilted interference fringes, achieving axial localization precision on the nanometer scale and allowing imaging of up to several tens of micrometers in depth within biological samples.
“MINFLUX and MINSTED have ... shown that there is physics to achieve one nanometer and better of localization precision for fluorescent molecules,” says scientist Steffen J. Sahl.
Finally, Hell and his colleagues introduced MINSTED in 2021. In this STED-SMLM mashup, the donut-shaped excitation beam of MINFLUX is replaced with a donut-shaped STED beam. Fluorophores appear brightest when centered in the middle of the donut hole. The team further optimized the technique in 2022, reducing the power of the STED beam and boosting spatial resolution still further. MINSTED experiments reported in the group’s latest study, published in November in Nature Biotechnology, measured the location of single molecules with precisions down to 4.7 Å—just under half a nanometer, or the scale of a single amino acid.
“MINFLUX and MINSTED have already shown that there is physics to achieve one nanometer and better of localization precision for fluorescent molecules,” maintains Sahl. “In the coming few years, I predict that any perceived last barriers of resolution at the molecular length scale will be shattered.”
Leveraging sheets of light
A third category of super-resolution microscopy is structured-illumination microscopy (SIM), pioneered by the late Mats Gustafsson in 2000. In SIM, interference between two or more beams forms a sinusoidal pattern used to illuminate the sample. The known illumination pattern and the unknown sample structure give rise to Moiré interference patterns, which have a lower frequency than their component frequencies. The patterns are then imaged by the microscope.
SIM indeed surpasses the Abbe limit, but unlike STED or SMLM, the resolution enhancement is inherently limited to a factor of two, with lateral resolutions typically hovering around 100 nm. What SIM does have on its side, however, is high temporal resolution, ease of use and compatibility with most fluorophores.
“SIM is still a very popular method compared to others, because while it doesn’t have the highest spatial resolution, it forms a good compromise between spatial and temporal resolution,” said Reto Fiolka, with the department of cell biology at UT Southwestern Medical Center, USA. “There have been studies where organelle dynamics have been studied at video rate (24 frames per second) or at even faster frame rates, which would be really challenging for STED or localization microscopy.”
SIM is also reasonably gentle imaging live-cell samples compared with its higher-resolution counterparts. When going fast, however, it still requires prolonged exposure to relatively high light intensities, since multiple images with different orientations and phases of the illumination pattern are needed to reconstruct the final super-resolution image. Researchers including Fiolka, a former postdoc in Gustafsson’s laboratory, have been searching for ways to lower the risks of phototoxicity and photobleaching in SIM.
OPSIM: Higher speed, lower phototoxicity
In 2022, Fiolka and colleagues reported on an innovative approach called oblique-plane SIM (OPSIM) that they believe has the potential to expand super-resolution microscopy to rapid, live-cell imaging. The technique combines SIM with light-sheet fluorescence microscopy (LSFM), which uses a thin sheet of light to excite only fluorophores within the focal volume. The entire sample is selectively illuminated only once per 3D image and thus exposed to three to five orders of magnitude less irradiation than in confocal microscopy.
“With a spinning disk or confocal microscope, but also with a SIM microscope, you can typically image only a few tens of image volumes before the sample is simply bleached,” said Fiolka. “In some cases, people have imaged [with LSFM] for days to weeks, acquiring thousands of image volumes.”
Also, because LSFM uses cameras for detection, millions of voxels can be acquired in parallel. Several tens to thousands of images can be recorded within only a few seconds, making it operable at much higher frame rates than either STED or SMLM.
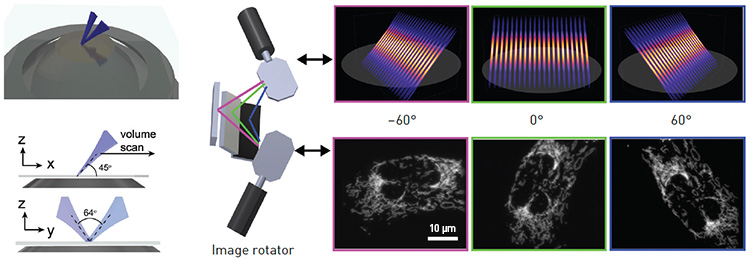 Left: In LSFM-SIM, two light sheets launched from a single objective lens overlap and create an interference pattern that is used for structured illumination. Right: An “image rotator” uses galvo mirrors to steer the structured light-sheet illumination, enabling multiple quick shots and keeping acquisition times low. [B. Chen et al., Nat. Meth. 19, 1419 (2022); reproduced with permission from Springer Nature]
Left: In LSFM-SIM, two light sheets launched from a single objective lens overlap and create an interference pattern that is used for structured illumination. Right: An “image rotator” uses galvo mirrors to steer the structured light-sheet illumination, enabling multiple quick shots and keeping acquisition times low. [B. Chen et al., Nat. Meth. 19, 1419 (2022); reproduced with permission from Springer Nature]
LSFM’s downside is its moderate spatial resolution, with lateral resolution generally ranging from 250 nm to 1 µm. By combining it with SIM, OPSIM effectively doubles the resolution of LSFM to about 150 nm. Instead of using multiple objectives, which others have attempted in the past, Fiolka’s team developed a microscope that illuminates the sample with a structured light sheet emitted at an angle from a single objective. This light sheet can be rotated to realize the multidirectional illumination required for SIM. Thereby, sample access is unconstrained, as in a conventional inverted microscope.
“If one comes from the perspective of the light-sheet world—whether it’s about keeping the sample happy and being fast, for example, or being able to acquire more than one volume per second of the entire cell—then doubling the resolution seems quite valuable,” he said. Tests of OPSIM on fluorescent nanospheres, collagen fibers, a tissue slice, and cancer and cardiac cells confirmed its lower phototoxicity versus traditional spinning-disk confocal systems, and its volumetric acquisition speed exceeding 1 Hz.
“Classic interference-based SIM approaches continue to surprise when it comes to biological application and contributing to scientific discoveries,” notes Schermelleh, “which highlights that it is the ‘biocompatibility’ of a technique that often makes the difference.”
The promise of deep learning
Deep learning—a type of machine learning based on artificial neural networks—has become a powerful tool for image processing and analysis, including in microscopy (see “Toward a Thinking Microscope,” OPN, July/August 2018). And super-resolution microscopy is no exception. Indeed, Schermelleh believes that increasing implementation of AI will make super-resolution microscopy “more user-friendly, robust, automated and quantitative,” allowing it to “harvest a much greater wealth of information in a lot more economic way.”
“A huge thing right now is the computational side of super-resolution microscopy—in particular, the development of processing approaches to enhance images using deep learning,” says Christophe Leterrier, leader of the NeuroCyto team at Aix Marseille University in France. He observes that incorporating deep learning into some standard super-resolution processing pipelines is allowing researchers to get more information out with less light, paving the way for imaging that’s gentler on biosamples.
Leterrier works closely with Ricardo Henriques and other colleagues on ZeroCostDL4Mic, an entry-level platform that aims to simplify and “democratize” deep learning for microscopy researchers. It leverages the free, cloud-based computational resources of Google Colab, a data analysis and machine-learning tool, to apply deep learning to tasks such as segmentation and denoising.
For super-resolution microscopy, researchers can use ZeroCostDL4Mic to employ Deep-STORM, a deep-learning network capable of reconstructing SMLM data from dense emitter datasets. Ideally, SMLM requires a sparsity of emitters, which translates to long acquisition times of seconds to minutes. Regions with a high density of overlapping emitters pose an algorithmic challenge, but Deep-STORM reportedly performs fast, precise image reconstruction with resolutions comparable to or better than existing methods, potentially allowing significantly shorter acquisition times.
In 2020, Li and colleagues developed a similar deep-learning-based reconstruction algorithm for high-density emitter data that they believe is better suited to live-cell imaging than Deep-STORM. Deep-residual-learning STORM (DRL-STORM) is an easy-to-use method that improves SMLM’s temporal resolution. DRL-STORM captured tubule structures in live mitochondria with greater clarity and less noise than Deep-STORM—and with a temporal resolution of 0.5 seconds.
Another application explored by Li and colleagues, using a deep-learning tool called a two-channel attention network (TCAN), seems almost like alchemy, apparently achieving the detail of super-resolution microscopy without all the extra effort. Li maintains that the TCAN-enabled resolution-enhancement algorithm the team developed “can transform [low-resolution] confocal images directly to super-resolution images, similar to or better than a STED image.”
After training with experimental data, the TCAN model reportedly maps the diffraction-limited input images into super-resolved ones without the use of special instrumentation or fluorophores. In a study published this year in the new journal PhotoniX, the researchers validated TCAN on diverse biological structures and dual-color confocal images of actin microtubules, improving the resolution from 230 nm to 110 nm. In addition, they demonstrated live-cell super-resolution imaging by revealing the detailed structures and dynamic instability of microtubules.
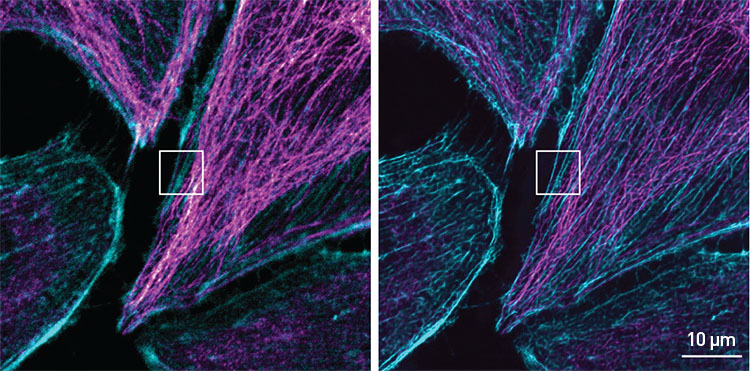 Confocal microscope input (left) and neural-network output (right) for microscopic image of actin microtubules as rendered by TCAN, a deep-learning framework for enhancing image resolution. Actin microtubules are in magenta, filaments are cyan. [B. Huang et al., PhotoniX 4, 2 (2023); CC-BY 4.0]
Confocal microscope input (left) and neural-network output (right) for microscopic image of actin microtubules as rendered by TCAN, a deep-learning framework for enhancing image resolution. Actin microtubules are in magenta, filaments are cyan. [B. Huang et al., PhotoniX 4, 2 (2023); CC-BY 4.0]
Super-res meets cryoEM
In addition to improvements in spatial resolution, speed and live-cell friendliness, researchers are tackling other challenges in super-resolution microscopy, like imaging thicker samples or combining super-resolution microscopy with non-optical modalities.
In diffraction-limited microscopes, a small amount of aberration, due to both refractive-index variations in the sample and imperfections in the optical design, does not significantly affect image quality. Super-resolution microscopes, however, rely on precise optical performance, and even small deviations from perfection can quickly degrade resolution, signal and contrast. Thus, “image quality can be degraded focusing at depths of even a few micrometers, compared to the depths of tens to hundreds of micrometers in conventional methods,“ according to Martin Booth of the University of Oxford.
Booth’s solution is adaptive optics, which he and his colleagues have demonstrated can benefit image quality for SIM, SMLM and STED. He believes that, with greater simplification and cost reduction in adaptive-optics hardware, more researchers will be able to apply super-resolution methods to thicker specimens, such as tissues, rather than just isolated cells.
Moerner’s group, on the other hand, is working on an emerging method known as super-resolved cryogenic correlative light and electron microscopy (srCryoCLEM), which combines super-resolution microscopy with another Nobel Prize–winning technique, cryogenic electron microscopy (cryoEM). CryoEM combines the flash-freezing of biological samples and the use of computational imaging to enable 3D imaging of biological structures at atomic resolutions.
Super-resolution microscopy retains its ability to surprise, and it is pushing the capabilities of light far beyond what Abbe and his contemporaries could have imagined.
“CryoEM is a really fantastic technique with incredibly high resolution. But the problem is, there are no specific non-perturbative labeling strategies,” says Peter Dahlberg, with the SLAC National Accelerator Laboratory, USA. “That’s where the synergy comes in, because of course, one of the real hallmarks of super-resolution microscopy is its ability to specifically label biomolecules of interest.” While cryoEM and conventional diffraction-limited light microscopy have been used side by side for more than a decade, the techniques’ vastly different resolutions have limited what can be done. Although still in early stages, techniques such as srCryoCLEM could help pinpoint the positions of distinct biomolecules identified by cryoEM at the nanometer scale.
As the combination with cryoEM suggests, super-resolution microscopy retains its ability to surprise, with new techniques and tricks continually being developed. As a result, it’s pushing the capabilities of light far beyond what Abbe and his contemporaries could have imagined.
Meeri Kim is a freelance science journalist based in Los Angeles, CA, USA.
For references and resources, visit:optica-opn.org/link/0423-super-res.
